Using Gotoh mapping
Until version 7.6, this was the sam_g2p step, because we first mapped the reads with bowtie2, then applied the G2P algorithm to bowtie2’s SAM output. Some of the V3LOOP samples mapped very badly to the HIV references, so we decided to try using Gotoh pairwise mapping, as described in issue #370. We chose the HIV1-C-BR-JX140663-seed reference to align against, because that’s the one that most closely aligns to the reference used in our G2P library.
Minimum Gotoh alignment score
In issue #395, we found that samples with no HIV in them were still mapping some reads to V3LOOP, sometimes thousands of reads. When we plotted the alignment scores, it was clear that our minimum score was too low.
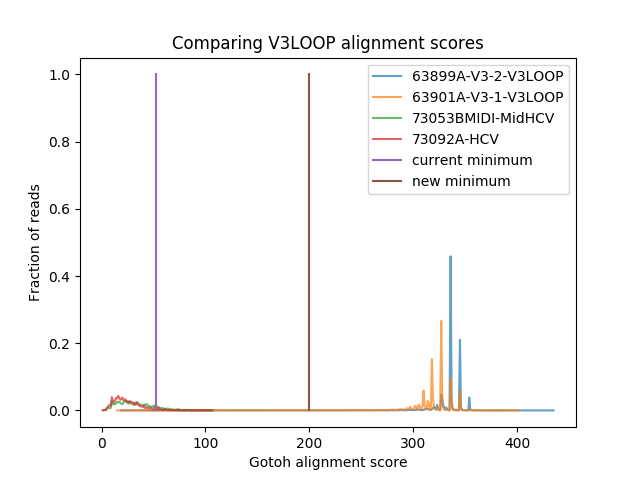
We changed the minimum score to be halfway between the two clusters of scores.